Have a look at the structures (a) and (b).
 |
 |
| (a) | (b) |
|---|
|
They could be thought of as mirror images of each other: one structure cannot be superposed on the other. Such molecules are known as enantiomers.
(+)/(−)
A pair of enantiomers often could be distinguished by their contrary optical activity, that is, rotation of plane-polarised light in opposite directions. Our (a) and (b) are two enantiomers of amphetamine: dextroamphetamine [also known as dexamfetamine, d-amphetamine, and (+)-amphetamine] and levoamphetamine [levamfetamine, l-amphetamine, (−)-amphetamine], respectively. In these names, ‘dextro’, ‘dex’, ‘d’ (all from the Latin word dexter, “right”) and ‘(+)’ refer to dextrorotation, i.e. right-hand or clockwise optical rotation, by this substance; and ‘levo’, ‘lev’, ‘l’ (from the Latin laevus, “left”) and ‘(−)’ indicate levorotation, i.e. left-hand or anticlockwise (counterclockwise) rotation*. The historical descriptors d and l could be easily confused with descriptors D and L; the former are deprecated by IUPAC [1].
A mixture containing equal amounts of a pair of enantiomers is called racemate. Such mixtures could be named using descriptors ‘rac’ or ‘(±)’; for example, an equimolar mixture of (a) and (b) is known as rac-amphetamine or (±)-amphetamine.
Descriptors such as ‘rac’ or ‘(±)’ refer to a physical property (absence of optical activity) observed in a macroscopic system, which could be a solution or a crystal but not a single molecule. Likewise, descriptors ‘(+)’, ‘(−)’, ‘d’, ‘l’, tell us that the system has optical activity. The system does not have to be a pure enantiomer to show dextrorotation: it could be a mixture with an excess of the ‘(+)’ isomer over the ‘(−)’ one.
R/S
Now let’s give systematic names to our structures. Amphetamine can be named substitutively 1-phenylpropan-2-amine. The structural difference between (a) and (b) lies in a spatial arrangement, or absolute configuration, of four different ligands, viz. hydrogen, amino group ‒NH2, benzyl group ‒CH2‒C6H5, and methyl ‒CH3, attached to the chiral carbon atom (C-2). To determine the configuration, the ligands are assigned priorities based on Cahn–Ingold–Prelog (CIP) sequence rules.
First, the atoms directly attached to the chiral centre are arranged in order of decreasing atomic number. In the structure (a), the order of preference is N > C = C > H. Next, as there are two carbon atoms bound to the chiral centre, we have to look at the precedence of atoms directly attached to them [3, P-92.2.1.1.2]. There are three hydrogen atoms in the methyl group: C(H,H,H), while the methylene carbon of the benzyl group is linked to two hydrogens and one carbon of the benzene ring: C(C,H,H). Since carbon is senior to hydrogen, C(C,H,H) > C(H,H,H), therefore, benzyl is senior to methyl. On the diagram (a), the chiral centre is positioned in such a way that the least-preferred ligand — in this case, hydrogen — points away from the viewer; the rest of the ligand sequence, N > Cbenzyl > Cmethyl, go anticlockwise. This configuration is indicated with the stereodescriptor ‘S’ (from the Latin sinister, “left”). This stereodescriptor is placed after the locant corresponding to the chiral atom, in our case, ‘2’, and enclosed in parentheses. Hence, the complete name of (a) will be (2S)-1-phenylpropan-2-amine. In (b), the ligands go in the opposite direction, i.e. clockwise, and the stereodescriptor ‘R’ (from the Latin rectus, “right”†), is used, thus (2R)-1-phenylpropan-2-amine.
Although the R/S convention is most commonly used for chiral carbon atoms, it could be as easily applied to any other tetrahedral chiral centres, including metal atoms [2, IR-9.3.4.2]. Moreover, the same symbols are employed to describe configuration of chiral trigonal pyramidal centres [2, IR-9.3.4.3; 3, P-93.3.3.2]. This is done by creating a tetrahedral centre by adding a “phantom atom” of the lowest priority to a trigonal pyramid. In compounds such as sulfoxides this phantom atom is placed in the site of a lone pair of electrons.
 |
 |
| (c) | (d) |
|---|
|
Consider (c) and (d), the enantiomers of sulforaphane. Its purely substitutive name is 1-isothiocyanato-4-(methanesulfinyl)butane, but I prefer the shorter functional class name, 4-isothiocyanatobutyl methyl sulfoxide. In the structure (c), the seniority order of the ligands to the chiral sulfur atom is O > C = C. Since C(C,H,H) > C(H,H,H), 4-isothiocyanatobutyl group is senior to methyl. On the diagram (c), the chiral centre is positioned so that the lone pair (or phantom atom) points away from the viewer; the sequence, O > C4-isothiocyanatobutyl > Cmethyl, go anticlockwise. Therefore, the complete name of (c) will be 4-isothiocyanatobutyl methyl (S)-sulfoxide, and that of its mirror image (d) 4-isothiocyanatobutyl methyl (R)-sulfoxide.
In contrast to macroscopic descriptors discussed above, ‘R’ and ‘S’ do not imply any optical activity or whatever other physical property: the “right” and “left” is just an arbitrary convention devised to distinguish between chiral centres.
D/L
What if the molecule has more than one chiral centre? No problem, the R/S convention allows us to specify the absolute confuguration at every one of them. For instance, the structure (e) could be systematically named (2R,3S,4R,5R)-2,3,4,5,6-pentahydroxyhexanal. However, nobody will call it that because there is a much better name: aldehydo-D-glucose.
 |
 |
| (e) | (f) |
|---|
|
The stereodescriptor ‘D’ here has the same origin as the now obsolete ‘d’, i.e. dexter. One of the historical names of D-glucose is ‘dextrose’ because its aqueous solution shows dextrorotation. The use of modern stereodescriptors ‘D’ and ‘L’ in the names of sugars, however, does not depend on the optical rotation. To understand what’s going on, we have to look at the simplest monosaccharide that has a chiral centre: glyceraldehyde.
 |
| (g) |
|---|
|
The structure (g) is that of D-glyceraldehyde, which also could be named substitutively (2R)-2,3-dihydroxypropanal. In carbohydrate nomenclature, a monosaccharide is given either ‘D’ or ‘L’ descriptor according to the configuration of the chiral atom furthest from the carbonyl group, which is known as the configurational atom [3, P-102.3.3]. In glucose, there are four chiral carbons: C-2, C-3, C-4 and C-5. We need to focus on C-5: if its configuration is the same as in D-glyceraldehyde — which is the case of the structure (e) — then the resulting name will also have ‘D’, as in aldehydo-D-glucose; otherwise, it is ‘L’.
What about the rest of the chiral atoms? Their relative configurations are implicit in the ‘gluco’ bit. In the names like aldehydo-D-gluco-hexose, ‘gluco’ is referred to as a “configurational prefix” [4, 2-Carb-4.3]. (We know by now that it is not a prefix but a combining form, that’s why I’ll keep the quotation marks). So instead of spelling out absolute configurations for all chiral centres using ‘R’ or ‘S’ descriptors, carbohydrate nomenclature specifies the absolute configuration of just one chiral centre (i.e. the configurational atom) using ‘D’ or ‘L’ while the relative configurations of other chiral centres are dealt with “configurational prefixes”. Resulting names are significantly shorter than substitutive ones; however, you need to look up what each “configurational prefix” means.
Apart from sugars, the stereodescriptors ‘D’ and ‘L’ are used in the nomenclature of cyclitols, amino acids and peptides [3, P-91.2.1.2.2].
C/A
There is no reason why the R/S convention cannot be used for polyhedra other than tetrahedron and trigonal pyramid. Indeed, the symbols ‘R’ and ‘S’ applied to octahedral systems are shown in earlier IUPAC recommendations [1, p. 29, diagrams (17) and (18)]. However, for the “other” polyhedra it’s currently recommended to use the chirality symbols ‘C’ (from “clockwise”) and ‘A’ (“anticlockwise”) [2, IR-9.3.4.4; 3, P-93.3.4.2]. These symbols are cited following the polyhedral symbol and configuration index.
Let’s see how this works on the example of the octahedral complex amminebromidochloridoiodidonitrito-κN-(pyridine)platinum(IV), or [PtBrClI(NH3)(NO2)(py)], which I shamelessly borrowed from the review of Constable [5, Fig. 14].
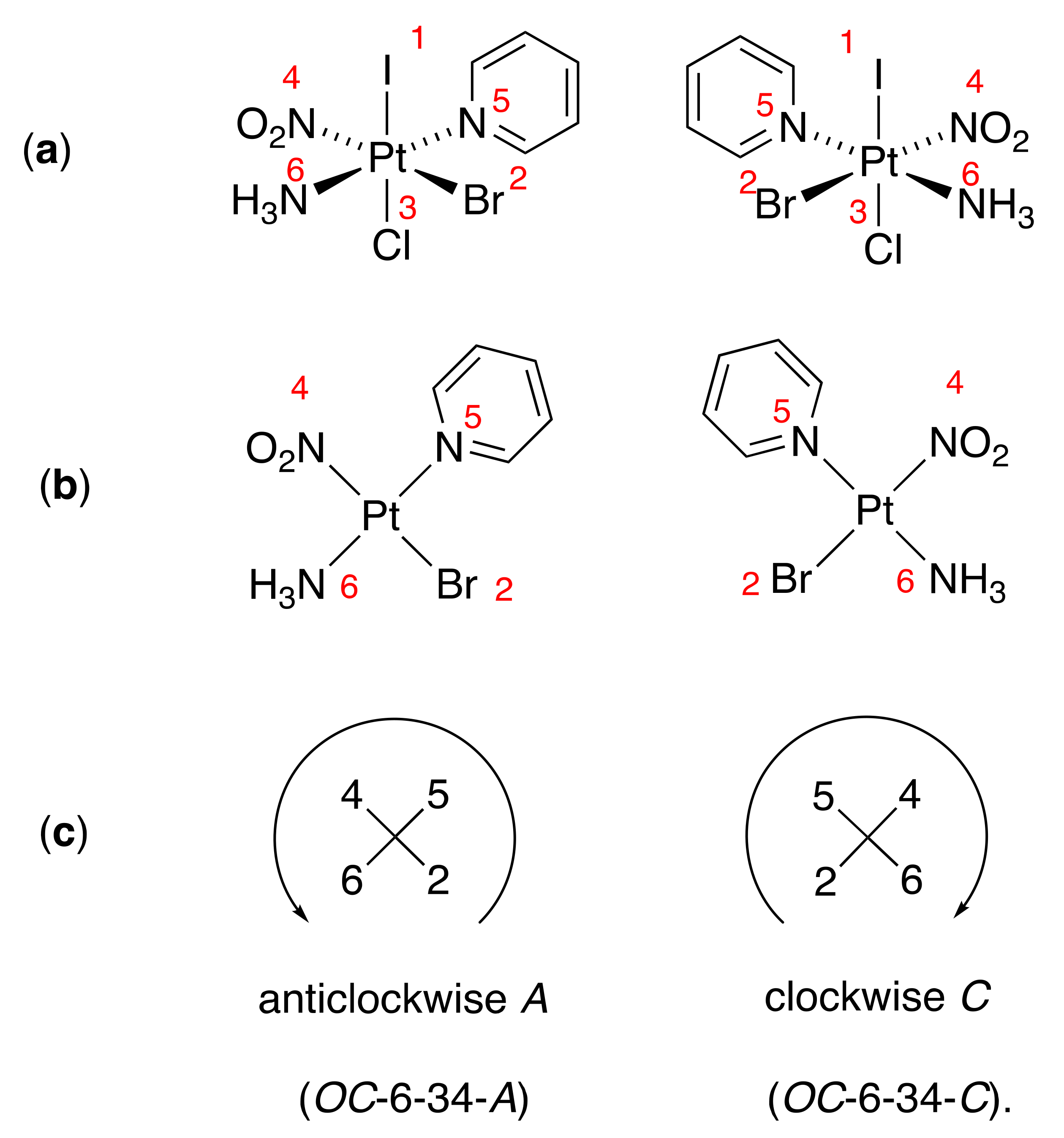
In line with the CIP rules, the priorities of the ligands are: ① (I), ② (Br), ③ (Cl), ④ (NO2), ⑤ (py) and ⑥ (NH3). The reference axis of the octahedron connects the highest priority ligand with the lowest priority ligand trans to it. In the case of [PtBrClI(NH3)(NO2)(py)], there is only one priority ① ligand, so the reference axis is ①—③ (I—Cl), and the first digit of the configuration index is ‘3’. The priority number of the ligand trans to the highest priority ligand in the plane that is perpendicular to the reference axis defines the second digit of the configuration index. In our case, NO2 is trans to Br, therefore the second digit of the configuration index is ‘4’. Now we look at the ligands in this plane from the side of priority ① ligand, i.e. iodine. We arrange the ligands in the numerical sequence starting from the highest priority atom. There are two possibilities, ②–⑤–④–⑥ and ②–⑥–④–⑤; since ⑤ > ⑥, the former sequence, as “the lowest numerical sequence”, is preferred‡. In the diagram on the left, the sequence ②–⑤–④–⑥ goes anticlockwise; accordingly, the complete descriptor for this structure will be ‘OC-6-34-A’ and the full name (OC-6-34-A)-amminebromidochloridoiodidonitrito-κN-(pyridine)platinum(IV). In the structure on the right, the sequence ②–⑤–④–⑥ goes clockwise, thus (OC-6-34-C)-amminebromidochloridoiodidonitrito-κN-(pyridine)platinum(IV).
Even though the R/S and C/A conventions deal with the same concept — viz. that of clockwise or anticlockwise arrangement of the ligands at the chiral centre according to the CIP rules — their practical uses differ significantly. The necessity to use the polyhedral symbol and configuration index in addition to the ‘C’ or ‘A’ descriptor effectively restrict the use of the C/A convention to the mononuclear coordination entities. Imagine how cumbersome the name like (2R,3S,4R,5R)-2,3,4,5,6-pentahydroxyhexanal would become if we had to specify that every chiral atom in it is T-4!
Δ/Λ
In the specific case of octahedral complexes containing bidentate ligands, another system, known as the skew-lines convention, is applied [2, IR-9.3.4.11].
3%5D2+.png) |
3%5D2+.png) |
| (h) | (i) |
|---|
|
The skew-lines are defined by the donor atoms of the ligands. In the case of structures (h) and (i), the enantiomers of tris(1,10-phenanthroline-κ2N1,N10)ruthenium(2+), the donor atoms are nitrogens, so we have three N—N lines. If you play with a 3-D structure of (e), you’ll discover that, no matter what you do, if you make any of the two N—N lines visually cross, with the intersection point more or less in the centre of the lines, the upper one always appears to be rotated approximately 60° anticlockwise relative to the lower one. This arrangement could be thought of as a left-handed helix and is assigned the Λ (lambda) descriptor, from the Ancient Greek λαιός, “left-handed”. The complete name of (h) will be Λ-tris(1,10-phenanthroline-κ2N1,N10)ruthenium(2+). On the contrary, with its enantiomer, the upper of any two intersecting N—N lines always appears to be rotated clockwise relative to the lower one. This isomer is assigned Δ (delta) descriptor, from the Ancient Greek δεξιός, “right-handed”, thus Δ-tris(1,10-phenanthroline-κ2N1,N10)ruthenium(2+).
Constable (2021) notes that using the C/A convention for tris(bidentate) complexes brings about the descriptors with different meaning of “clockwise” and “anticlockwise” compared to that of Δ and Λ [5, Fig. 16]. This is because all the donor atoms have the same priority and to differentiate them, the priming convention is used [2, IR-9.3.5.3]. The pairs of donor atoms in each bidentate ligand are arbitrarily unprimed, primed, or double primed. Since the priorities are ① > ①′ > ①″, the reference axis of the octahedron is ①—①″. The atoms left in the plane perpendicular to it have priorities ①, ①′, ①′ and ①″. The numerical sequence ①–①′–①′–①″ goes clockwise in Λ isomer and anticlockwise in Δ isomer, giving (OC-6-1″1′-C) for Λ and (OC-6-1″1′-A) for Δ. Confused? I bet you are. Personally, I prefer more tangible helicity of the Δ/Λ system to the arbitrary handedness of the C/A convention.
P/M
Some molecules do not have any chiral centres and yet are chiral. Observe (j) and (k), two enantiomers of hexahelicene:
 |
 |
| (j) | (k) |
|---|
|
You don’t need any seniority rules to see that the structure (j) is a right-handed helix and (k) is a left-handed helix. Their chirality is indicated by the stereodescriptors ‘P’ (“plus”) and ‘M’ (“minus”), respectively [3, P-92.1.2.2.1].
The descriptors ‘M’ and ‘P’ also could be used to specify chirality of compounds such as allenes and hindered biaryls, however in these cases you have to apply the CIP rules.
To sum up:
- The concepts of right- and left-handedness in chemical names are conveyed by a variety of conventions: (+)/(−), P/M (plus/minus), d/l, D/L (dexter/laevus), Δ/Λ (δεξιός/λαιός), R/S (rectus/sinister), C/A (clockwise/anticlockwise).
- Descriptors ‘(+)’, ‘(−)’, ‘d’, ‘l’, ‘rac’ or ‘(±)’ refer to optical activity in a macroscopic system but don’t tell us anything about molecular geometry.
- The descriptors ‘R’ and ‘S’ define the absolute configuration at tetrahedral and trigonal pyramidal chiral centres using the CIP sequence rules.
- The descriptors ‘C’ and ‘A’ define the absolute configuration at other polyhedral chiral centres using the CIP sequence rules.
- The descriptors ‘D’ and ‘L’ are assigned to the whole molecule according to the absolute configuration of one special chiral centre, aka configurational atom. The configurations of other centres relative to the configurational atom are defined by “configurational prefixes” such as gluco, erythro, threo, ribo, etc. This system is used in nomenclature of carbohydrates, amino acids and cyclitols.
- The descriptors ‘Δ’ and ‘Λ’ describe configurations of octahedral complexes containing bidentate ligands.
- The descriptors ‘M’ and ‘P’ describe configurations of molecules that lack chiral atoms.
Closing remarks
None of these symbols are particularly original. Italicised element symbols ‘C’, ‘P’ and ‘S’ are used in both inorganic and organic nomenclature [2, IR-2.9; 3, P-16.6.2]. The symbol ‘Δ’ is used in organic nomenclature for localised double bonds [3, P-25.7.1.2] and still widely employed by biochemists to indicate unsaturation in lipids. The capital ‘D’ (although not the small cap ‘D’, but honestly, how many people see the difference?) is a symbol for deuterium. If I were to propose a new stereochemical notation now, I would go for ‘↺’ (anticlockwise open circle arrow, Unicode U+21BA) and ‘↻’ (clockwise open circle arrow, Unicode U+21BB). That’s how our names would look like:
-
↻-glyceraldehyde (g)
(2↻,3↺,4↻,5↻)-2,3,4,5,6-pentahydroxyhexanal (e)
(OC-6-34-↻)-amminebromidochloridoiodidonitrito-κN-(pyridine)platinum(IV)
↺-[Ru(phen)3]2+ (h)
↻-hexahelicene (j)
Beautiful.
| * | Of course, “right” is not the same as “clockwise”. Clock hands go from left to right only half of the time, from 9 to 3 o’clock (and right to left the other half of the time). Likewise, if the plane of polarisation rotates clockwise, it’s dextrorotating only half of the time. The chirality rules really rely on concepts of “clockwise” and “anticlockwise”, not “right” and “left”. It’s unfortunate that the confusion lives in chemical nomenclature. |
| † | On top of the confusion between “right” and “clockwise”, the descriptor ‘R’ is based on wrong etymology. The Latin word rectus means “right” but in a sense “straight”, “upright”, i.e. “not curved”. It does not imply the “right-hand” direction at all. Thus rectus is a different “right” compared to dexter, which indeed is the opposite of “left”. |
| ‡ | “The lowest numerical sequence is that having the lower number at the first point of difference when the numbers are compared digit by digit from one end to the other” [2, IR-9.3.3.6]. |
References
- Cross, L.C. and Klyne, W. (1976) Nomenclature of Organic Chemistry. Section E: Stereochemistry (Recommendations 1974). Pure and Applied Chemistry 45, 11—30.
- Connelly, N.G., Hartshorn R.M., Damhus, T. and Hutton, A.T. Nomenclature of Inorganic Chemistry: IUPAC Recommendations 2005. Royal Society of Chemistry, Cambridge, 2005.
- Favre, H.A. and Powell, W.H. Nomenclature of Organic Chemistry: IUPAC Recommendations 2013 and Preferred IUPAC Names. Royal Society of Chemistry, Cambridge, 2014.
- McNaught, A.D. (1996) Nomenclature of carbohydrates (IUPAC recommendations 1996). Pure and Applied Chemistry 68, 1919—2008.
- Constable, E.C. (2021) Through a glass darkly — Some thoughts on symmetry and chemistry. Symmetry 13, 1891.







No comments:
Post a Comment