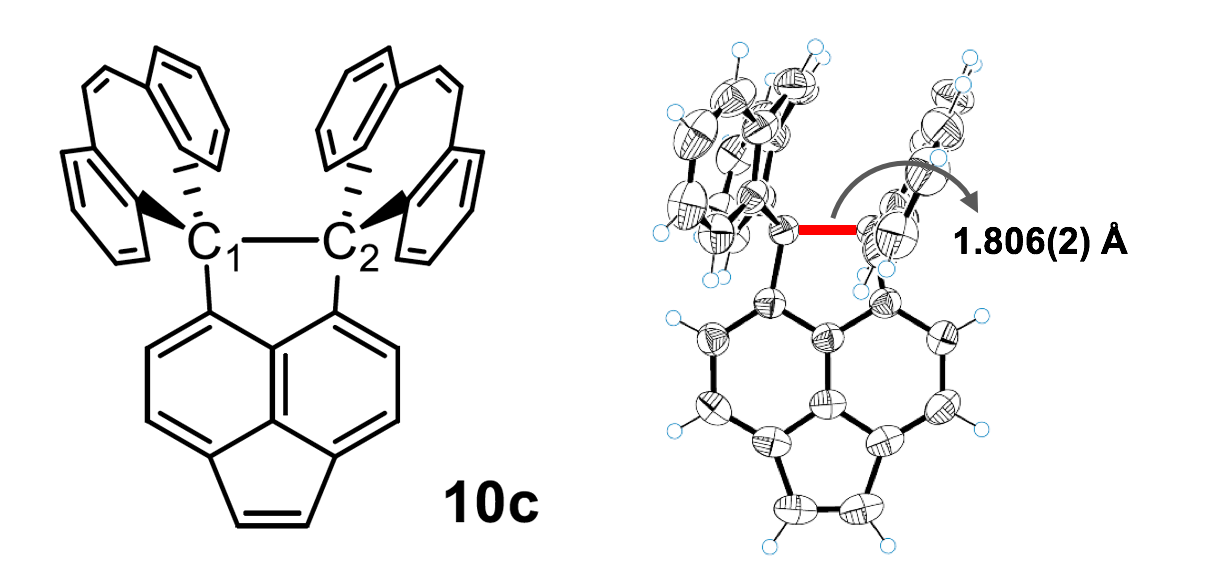
While I wasn’t looking, the InChI folks implemented the metal-reconnected layer. Isn’t it nice? I discovered it quite by chance thanks to the Beilstein-Institut ChemInfo Labs page. You can see how it works on InChI Web Demo.
Consider ferrocyanide (a):
 |
| (a) |
|---|
|
Its standard InChI is:
| InChI=1S/6CN.Fe/c6*1-2;/q;;;;;;-4 | (1) |
The main layer contains two types of entities: 6CN (i.e. six CN molecules) and Fe (one iron atom). If we try to convert (1) back to structure, using the same Web Demo tool, we get six free-floating CN• radicals and a separate Fe4− anion. Ew. But if we tick the “Include Bonds to Metal” box in the Web Demo tool, we have
| InChI=1/6CN.Fe/c6*1-2;/q;;;;;;-4/rC6FeN6/c8-1-7(2-9,3-10,4-11,5-12)6-13/q-4 | (2) |
where the metal-reconnected layer (/r) appears. It looks like an alternative InChI added directly after the standard one, with its own connectivity (/c) and charge (/q) sublayers. In this layer, there is only one entity: C6FeN6, i.e. [Fe(CN)6]. The string (2) is correctly converted back to the structure (a).
Now let’s look at the structure of a salt known as Prussian Blue (b):
 |
| (b) |
|---|
|
Its standard InChI is:
| InChI=1S/18CN.7Fe/c18*1-2;;;;;;;/q;;;;;;;;;;;;;;;;;;3*-4;4*+3 | (3) |
The main layer contains two types of entities: 18CN (i.e. 18 CN molecules) and 7Fe (seven iron atoms). Converting (3) to structure brings about a horrible mess. With “bonds to metal”, however, we get
| InChI=1/18CN.7Fe/c18*1-2;;;;;;;/q;;;;;;;;;;;;;;;;;;3*-4;4*+3/r3C6FeN6.4Fe/c3*8-1-7(2-9,3-10,4-11,5-12)6-13;;;;/q3*-4;4*+3 | (4) |
In the metal-reconnected layer (/r) we see two different types of entities: 3C6FeN6 (i.e. three [Fe(CN)6]) and 4Fe. The string (4) is correctly converted back to the structure (b).
Years ago, I was complaining (to the universe) about different InChIs for the same molecular entity, viz. chromate (c—e). I’ve revisited it with the new version of InChI.
![[Cr(O)2(O-)2]](https://blogger.googleusercontent.com/img/b/R29vZ2xl/AVvXsEiMl79HNr0UpiyYYGh19PXfrVS8tgjZpGFAXZVCc4UOf9mkbRi22X04ihTqlc4kIt3MhjkoadFSu1o4S_bm2B_MrMmvKwlJB__guvw6LTHk2KhQpN1daOMI2lDyeUkHefZXhMy2pw/s320/%5BCr(O)2(O-)2%5D.png) |
![[Cr(O)4]2-](https://blogger.googleusercontent.com/img/b/R29vZ2xl/AVvXsEig_6nDbBjkdqedyqMZqcaqsMjAKQTNTh6qfYC695EGI2cTTKtyAbJh13mjeVrv_7wLrr6mcW1FMJVxrR1n9JCZHH9R5Dwb-gWMGpJSDl2_VwEirZWvOGQv359HmZuN3mLmDDS19A/s320/%5BCr(O)4%5D2-.png) |
![[Cr(2+)(O-)4]](https://blogger.googleusercontent.com/img/b/R29vZ2xl/AVvXsEhcX_-K6YsWcufdFVEykYISID9dKdDFxNYAtXgcZvGq4cOeyXrwgE4DhOAAQVRu23tiP0soKyY7s05pGIabeMwIZX1YoPhL73RGw6zuauvpkJ9w5DQMnLqzS9PTzcO0tsKc1yoYVg/s320/%5BCr(2+)(O-)4%5D.png) |
| (c) | (d) | (e) |
|---|
chromate (trivial) tetraoxidochromate(2−) (additive) tetraoxidochromate(VI) (additive) |
Alas, the standard InChIs for the representations (c), (d) and (e) remain different. Try to convert them back to structures: they also are all different and all wrong (all have extra hydrons). Nevertheless, I see a sign of progress: the metal-reconnected layers for the corresponding strings (5), (6) and (7) are identical!
| (c) | InChI=1/Cr.4O/q;;;2*-1/rCrO4/c2-1(3,4)5/q-2 | (5) |
| (d) | InChI=1/Cr.4O/q-2;;;;/rCrO4/c2-1(3,4)5/q-2 | (6) |
| (e) | InChI=1/Cr.4O/q+2;4*-1/rCrO4/c2-1(3,4)5/q-2 | (7) |
Moreover, all three strings, (5), (6) and (7), are converted back to the structure (c).
What about our old friend ferrocene? Depends how you draw it. I’ll stick to the ChEBI’s decacoordinate-iron representation (f):
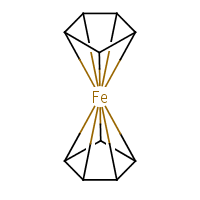 |
| (f) |
|---|
|
The standard InChI for ferrocene is:
| InChI=1S/2C5H5.Fe/c2*1-2-4-5-3-1;/h2*1-5H; | (8) |
Converting (8) to structure results in two standalone cyclopentadienyl radicals and a neutral iron atom. With “bonds to metal”:
| InChI=1/2C5H5.Fe/c2*1-2-4-5-3-1;/h2*1-5H;/rC10H10Fe/c1-2-4-5-3(1)11(1,2,4,5)6-7(11)9(11)10(11)8(6)11/h1-10H | (9) |
In the /r layer we see a single entity, C10H10Fe, i.e. [Fe(C5H5)2]. The string (9) is correctly converted back to the structure (f).